Eritropoiesi
Tutta la parte cellulare del sangue circolante è prodotta dal midollo osseo rosso la cui massa è pari a circa il 2% del peso corporeo. La cellula che produce le singole filiere si chiama cellula staminale ed ha la caratteristica che la sua duplicazione produce una cellula destinata alla differenziazione in una delle tre filiere ed una cellula destinata a rimanere staminale. Normalmente il midollo rosso produce ogni giorno 350 miliardi di cellule per ogni chilo del suo peso; poiché le cellule che sono prodotte rappresentano il 90% circa di quelle immesse nelle filiere dalle cellule staminali la quantità di cellule globali nel midollo rosso si aggira intorno ai 400 miliardi per ogni chilo.
Il midollo rosso produce circa 2.5 miliardi di globuli rossi per Kg di peso corporeo al giorno. L’attivià formativa è svolta all’interno di nicchie dello stroma midollare in cui sono annidati i precursori immaturi. La cellula staminale totipotente (SC) è ospitata nel cosiddetto “compartimento LSK” dove LSK è un acronimo per (lineage-/Scal+/c-kit+), dove è possibile riscontrare tre tipi di SC: le LT-SC, caratterizzate da una notevole capacità di auto-rinnovamento, una forte espressione dell’endoglina e del CD150, ma con bassa espressività per il Thy1.1, Rho e soprattutto del CD34; le ST-SC, caratterizzate da una ridotta capacità di autorinnovamento e dalla forte espressione del fenotipo CD34; e le MPP o progenitori megacariocitico/eritrocitico, in cui spicca, oltre alla positività per il CD34, quella per il determinante di membrana FLT3, la negativizzazione del Thy1-1 e la bassa espressione del CD11b. Ogni SC può andare incontro a due possibili esiti: un auto-rinnovamento simmetrico in cui da una cellula ne derivano due figlie che rimangono come tali, oppure un auto-rinnovamento asimmetrico in cui delle due cellule figlie una rimane SC, una diventa MPP. Le SC hanno un potenziale biologico estremamente elevato: una sola di esse è sufficiente a ricostituire un midollo normale nel ratto. L’auto-rinnovamento e il commissionamento sono il risultato dell’interazione di quattro vie regolatrici principali. Il complesso dell’ SCF, formato dal ligando SCF di origine fibroblastica ed endoteliale e dal suo recettore specifico, prodotto del proto-oncogene c-kit (CD117), la combinazione dei quali comporta l’autofosforilazione del recettore e successiva trascrizione dei geni che codificano per l’homing delle SC nelle nicchie mielopoietiche e l’auto-mantenimento delle medesime.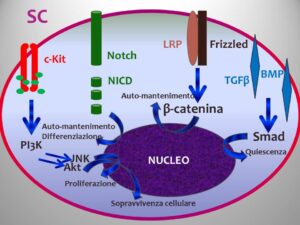
Il complesso formato dal recettore Notch e i suoi ligandi Delta1-3 e Jagged1-2 di origine osteoblastica, che attivando una gamma-secretasi porta al rilascio della porzione intracellulare del recettore Notch, la quale migra nel nucleo, dove complessandosi con il fattore di trascrizione CSL e i cofattori MAML induce l’attivazione della trascrizione sia dei geni coinvolti nel mantenere lo stato di cellula staminale indifferenziata in collaborazione con il complesso Wnt, sia dei geni che promuovono la differenziazione, soprattutto delle MPP. Il complesso del ligando Wnt, di origine stromale ma proveniente anche dalle SC CD34+ (azione autocrina), che combinandosi con il recettore Frizzled porta alla complessazione dello stesso con una proteina di membrana correlata al recettore lipoproteico e all’attivazione di una via interna che annovera la β-catenina, la cui complessazione con il fattore di trascrizione TCF induce un aumento della trascrizione dei geni coinvolti nell’auto-mantenimento. Ed il complesso Smad, a sua volta distinguibile in due vie ad effetto opposto: la via mediata dai ligandi della famiglia del TGFβ, che complessando il suo recettore omonimo attiva Smad2 e Smad3 con conseguente aumento di trascrizione di geni che mantengono la quiescenza delle SC e la via mediata dai ligandi BMP, che, attivando Smad5, sembra importante per l’auto-rinnovamento durante la mielopoiesi embrionale [1].
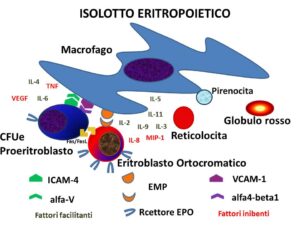
La cellula MPP che va incontro ad una reduplicazione asimmetrica, nella componente destinata ad ulteriori passi maturativi, diventa CMP o progenitore mieloide comune, il quale si differenzia in una cellula orientata in senso granulocito-monocito-poietico (GMP) o in una cellula orientata in senso Eritro-trombocitario (MEP). A questo punto la MEP, che è la cellula che qui interessa, lascia il compartimento dei progenitori multipotenti per entrare nel compartimento dei progenitori commissionati come BFU-E. L’attività proliferativa e differenziativa delle BFU-E è regolata in piccola parte da un ormone di origine renale, l’eritropoietina (EPO) e in gran parte da citochine prodotte in loco dalle cellule dello stroma, ma anche provenienti dal circolo sistemico: hanno azione facilitante le interleuchine 3, 4, 5, 6, 9 e 11, hanno azione bloccante l’interleuchina 8, il MIP-1, il VEGF e i TNF. Le BFU-E si differenziano in CFU-E, sulle quali il numero dei recettori per l’EPO aumenta e compare il recettore per la transferrina (TfR1): ciò vuol dire che in questo tipo di cellula è iniziata la produzione di emoglobina ed è iniziata, quindi, l’assunzione sistematica, continua e preferenziale di Fe. Anche le CFU-E sono soggette all’azione facilitante di alcune interleuchine (2 e 6) e ostacolante del TNFα, ma in minor misura rispetto alle BFU-E. Le CFU-E si differenziano in proeritroblasto, una cellula di circa 30 μm di diametro, in cui l’aumento progressivo dell’emoglobina intracellulare, la graduale riduzione del volume e il raddoppio del numero delle cellule in ogni fase del processo maturativo lo portano a diventare prima eritroblasto basofilo, poi eritroblasto policromatofilo e quindi eritroblasto ortocromatico. Durante questo processo le cellule conservano la capacità riproduttiva fino all’ultima tappa, quando si realizza la degenerazione e l’espulsione del nucleo. Ogni CFU-E, quindi, genera 2 proeritroblasti, 4 eritroblasti basofili, 8 eritroblasti policromatofili, 16 eritroblasti ortocromatici, i quali, a loro volta, esitano in 32 reticolociti [2].
Questa maturazione si compie nel giro di circa 5 giorni e si conclude con la produzione di reticolociti, cioè di una cellula anucleata di circa 7.5 μm di diametro, granulata, con un volume che è pari al 25% di quello del proeritroblasto. Il reticolocita matura definitivamente in globulo rosso nel giro di 24 h, in massima parte nel midollo, in parte, molto piccola, in circolo, dove finisce per rappresentare circa l’1% dei globuli rossi nel sangue.
Le tappe finali dall’eritroblasto ortocromatico al reticolocita maturo sono costituite essenzialmente dalla rapida eliminazione dei recettori TfR1, dalla enucleazione con produzione di un pirenocita, formato dal nucleo degradato dell’eritroblasto per un processo di deacetilazione e in minor misura di demetilazione degli istoni, dall’eliminazione dei mitocondri attraverso l’attivazione del fattore NIX , proteina del complesso BCL2 e in minor misura per autofagia tramite alcune serin-treonin-kinasi (ULK1 e Atg7), nonché dalla distruzione dei ribosomi per opera delle ribonulceasi [3].
Non tutte le cellule anucleate, comunque, diventano reticolociti maturi e, quindi, globuli rossi, perché il 10% è fagocitato per vari motivi strutturali all’interno del midollo stesso. Il centro motore di questa lunga filiera maturativa è il cosiddetto “isolotto eritropoietico”, formato da uno o più macrofagi, che costituiscono la componente cellulare centrale deputata sia alla produzione delle citochine, sia al controllo del metabolismo del Fe, sia alla fagocitosi dei nuclei e delle cellule difettose. In linea di massima tutte le cellule fino allo stadio eritroblasto ortocromatico rimangono adese alla membrana dei macrofagi tramite alcune proteine di contatto ivi situate, quali VCAM-1, EMP e alfa-V, che trovano sulle cellule in fase maturativa le corrispondenti molecole alfa1-beta1, EMP e ICAM-4 [4].
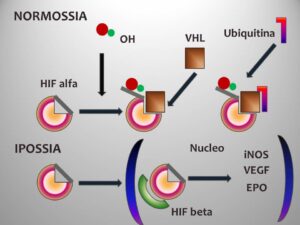
Da un punto di vista strettamente umorale, quindi, il ruolo chiave dell’eritropoiesi è sostenuto dall’EPO, prodotta da cellule epitelioidi dei capillari della corteccia renale. In condizioni di normossia gli HIFs, o fattori indotti dall’ipossia, sono mantenuti allo stato ridotto dall’HIF-prolil-idrossilasi e, poi, complessati alla VHL E3, una ubiquitina, sono incamerati nei proteasomi e indirizzati alla eliminazione. In condizioni di ipossia, l’aumento del succinato all’interno dei mitocondri inibisce l’idrossilazione degli HIF, permettendo loro di agire. L’HIF-1, complessato agli HREs, induce la sintesi degli enzimi della glicolisi, la sintesi del VEGF e dell’EPO. L’EPO, una volta legato il recettore specifico, blocca l’azione della combinazione del Fas/Fas ligando tra eritroblasti ortocromatici ed eritroblasti più immaturi, nonché attiva il JAK2, con conseguente attivazione della PI3-chinasi e della STAT5, ambedue facilitatori della trascrizione genica, che nel caso specifico riguarda geni coinvolti nella crescita e nella maturazione degli eritroblasti; infine l’EPO riduce la sintesi di epcidina1, che vedremo essere l’inibitore principale della mobilizzazione del Fe [5] .
L’evento molecolare fondamentale nella maturazione del globulo rosso è la sintesi dell’emoglobina e cioè delle catene proteiche che nell’adulto e nelle varie fasi dello sviluppo embriofetale caratterizzano questa molecola e dell’eme.
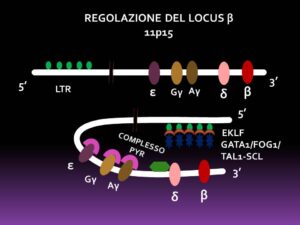
La parte proteica dell’emoglobina, cioè le due doppie eliche che formano la globina, sono sintetizzate a partire dai geni specifici, tramite una regione del cromosoma 11 o 16, specifica, situata qualche decina di basi verso l’estremità 5’, formata da 200-400 bp ad alta sensibilità per la DNAse I ( DNAse I HS), definita LCR (Locus Control Region). Questa sequenza di basi lega diversi tipi di proteine trascrizionali: GATA-1, specifico per i box (A/T)GATA(A/G) con l’indispensabile collaborazione sia della proteina FOG-1 che del complesso TAL-1/SCL, fattore di trascrizione helix-loop-helix, EKLF o erythroid Kruppel-like factor specifico per i box CACC, il fattore YY-1 e l’SP1. Tutte queste proteine hanno la funzione di formare uno o più complessi promotori, costringendo l’estremità LCR a ripiegarsi verso l’estremità 3’ e ad assumere l’aspetto di un’ansa con la parte centrale occupata dai DNAse I HS, dove, appunto, si localizzano i fattori trascrizionali.Contemporaneamente, nell’adulto, il complesso PYR provvede a sopprimere la trascrizione dei geni ε, γ e δ, in modo che l’effetto promotore si concentri esclusivamente sul gene β. In questo modo gli LCR hanno la funzione di aumentare la trascrizione dei geni verso l’estremità 3’, isolando il locus da trascrivere dalla cromatina vicina e aprendone la cromatina. FOG-1 avrebbe il compito principale di rimuovere GATA-2 dai box GATA. Il GATA-2, infatti, è il principale fattore di repressione trascrizionale dei geni dell’α- e β-globina, il cui equilibrio con GATA-1 è governato soprattutto dal TNFα, che determina l’aumento trascrizionale di GATA-2 [6].
L’RNAm frutto della trascrizione è molto stabile, avendo un’half life di 10-20 h. Questa stabilità è sostenuta da due principali meccanismi di controllo del trascritto: dai 3’UTR degli RNAm dell’α- e β-globina, distinti strutturalmente e funzionalmente, nonché, soprattutto, dal complesso NMD o nonsense-mediated mRNA decay, il quale entra in funzione quando i ribosomi in fase di esplorazione dell’mRNA dopo lo splicing post-traslazionale individua una sequenza troncata prematuramente (PTC) oltre i 50-54 nucleotidi verso 5’ dalla giunzione di due esoni. In questo caso la proteina CBP80, situata all’estremità 5’ dell’mRNA, si pone sopra PTC ripiegando il tratto corrispondente di mRNA. Il successivo decapping e rimozione esonucleotidica prima nella direzione 3’-5’ poi in quella 5’-3’ ad opera degli esosomi, porta alla degradazione completa dell’mRNA danneggiato [7]. Ma esiste anche una stabilizzazione post-traduzionale, soprattutto per quanto riguarda le catene α della globina, prodotta da una proteina specifica, AHSP, la cui sintesi è promossa dai fattori di trascrizione GATA-1 e Oct-1.
La sintesi dell’eme inizia all’interno dei mitocondri ad opera dell’enzima principale dell’intero processo che è l’ALA-sintetasi, per mezzo della quale da una molecola di glicina e da una di succinil-CoA è prodotta una molecola di Acido-d-aminolevulinico, il quale, una volta uscito dal mitocondrio, subisce nelle cisterne del reticolo endoplasmatico liscio una ciclizzazione a Porfobilinogeno; quattro di queste molecole sono poi condensate ad opera di una deaminasi e ciclizzate a formare Uroporfirinogeno III, che per decarbossilazione è trasformato in Coproporfirinogeno III. Da questa molecola, rientrata nel mitocondrio dove subisce una doppia ossidazione, deriva la Protoporfirina IX, cioè un anello tetrapirrolico. Sarà la ferrochelatasi che, infine, inserirà il Fe++ al suo interno. La sintesi degli enzimi di questo ciclo, nell’eritroblasto, a differenza che in altre cellule, è molto stabile ed è influenzata soprattutto dalla disponibilità di Fe, la cui riduzione blocca proporzionalmente la trascrizione del gene che codifica l’ALA-sintetasi [8] .
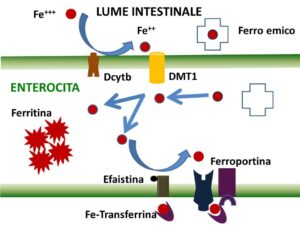
L’altro elemento indispensabile per costruire l’eme è il Fe. Una dieta equilibrata di un adulto ha un contenuto medio di Fe di 20 mg/24 h. Di tutto il Fe che si ingerisce è assorbito poco meno del 30%, equamente diviso fra Fe minerale e Fe emico. In condizioni di particolare fabbisogno questa quota può aumentare fino al 35%. Il Fe minerale è assorbito generalmente dopo complessazione con acidi organici mentre il Fe emico è assorbito come tale. Le cellule della mucosa intestinale, soprattutto del duodeno e del primo tratto del digiuno, assorbono il Fe inorganico veicolandolo all’interno della cellula per mezzo della DMT1, dopo averlo ridotto a Fe++ ad opera di un enzima specifico, il Dcytb. Al termine del processo risulta assorbito soltanto l’1.5% del quantitativo ingerito. Il Fe emico, invece, è assorbito forse tramite mediatori specifici ed è liberato dal complesso con l’eme all’interno dell’enterocita, dove raggiunge una concentrazione pari ad ¼ della quantità ingerita. La cellula intestinale, dopo aver ossidato a Fe+++ tutto il Fe assorbito, ne conserva una parte nella ferritina propria, che non recupererà mai più dopo i suoi 3-4 giorni di vita, e libera il resto nel sangue tramite l’azione della ferroportina insieme all’ efaistina, un enzima ossidasico specifico rame-dipendente. Questo ciclo si realizza in poche ore e la velocità di assorbimento è equivalente alla velocità di cessione. E’ evidente che il contenuto di Fe del sangue portale, cioè del sangue venoso che dall’intestino giunge al fegato, è superiore a quello del resto del sangue circolante. A livello del sangue il Fe+++ è veicolato dalla transferrina, una proteina di trasporto prodotta dal fegato, ai tessuti periferici, per la maggior parte in ragione di 1 atomo per molecola. La transferrina si lega alle cellule che presentano sulla superficie il recettore specifico; quando ne prende contatto viene internalizzata sotto forma di vescicole, entro le quali l’aumento dell’acidità determina la liberazione del Fe+++ dalla sua proteina di trasporto, che viene riportata sulla membrana cellulare insieme al suo recettore, ambedue integri, mentre il Fe+++ è ridotto a Fe++. All’interno delle cellule dei tessuti periferici, compreso l’eritroblasto, il Fe è distribuito fra Fe immediatamente utilizzabile per la sintesi delle ferro-proteine e in Fe depositato nelle molecole di ferritina. In particolare, nei macrofagi la quantità di Fe ferritinico è molto più elevata del Fe metabolico, tanto che parte del Fe di deposito è accumulato in un polimero della ferritina, l’emosiderina: la differenza fra le due molecole è che la ferritina ospita 4500 atomi di Fe per molecola, corrispondenti a circa il 20% del suo peso secco, e si trova in circolo, mentre l’emosiderina contiene il 50% in più di Fe e non si trova in circolo. Il Fe ferritinico è mobilizzabile dai macrofagi e dagli epatociti in cui è depositato grazie sia alla ferroportina, come nell’enterocita, sia ad una proteina rame-dipendente, non enzimatica come l’efaistina, che è la ceruloplasmina [9].
La sideremia, cioè la concentrazione di Fe, nel sangue del neonato ammonta a 1.8 μg/mL, nel bambino a 0.8 μg/mL, nell’adulto maschio a 1.15 μg/mL e nell’adulto femmina a 1.05 μg/mL, saturando soltanto, in tutti i casi, il 30% della capacità totale di legare Fe da parte della transferrina. I livelli di sideremia sembrano seguire un andamento circadiano, poiché al risveglio raggiungono i valori più alti, mentre dopo 12 h toccano i valori più bassi. La ferritinemia fra i 6 mesi e i 12 anni di età si mantiene intorno ai 30 ng/mL, per stabilizzarsi a 66 ng/mL nella donna fino ai 45 anni per poi aumentare a 100 ng/mL dopo questa età, mentre negli uomini a 18 anni arriva in media a 46 ng/mL, a 35 anni a 170 ng/mL, a 45 anni a 196 ng/mL.
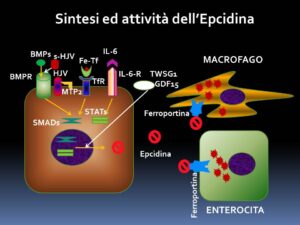
Il regolatore principale di questo complesso metabolismo è l’epcidina, un ormone sintetizzato dal fegato, che ha la funzione di complessare la ferroportina sia negli enterociti che nelle cellule periferiche, compresi i macrofagi, determinando la fosforilazione della proteina Jak2, bloccando, quindi, la mobilizzazione del Fe e inducendo la sintesi di ferritina. L’epcidina, a sua volta, è regolata, nella quantità in cui viene prodotta, dall’azione stimolante delle BMPs, dell’interleuchina 6, dell’HFE e della transferrina, mentre subisce l’azione inibente della Matriptasi 2, dell’HIF e di due citochine di origine midollare la GDF15, prodotta dagli ertitroblasti terminali e la TWSG1, sintetizzata, forse, dalle cellule più immature della filiera eritroide. In particolare, l’azione delle BMPs è fortemente influenzata dall’emojuvelina di membrana (mHJV) la cui inattivazione e trasformazione in emojuvelina solubile è sostenuta dalla Matriptasi 2 [10].
Nell’economia generale del metabolismo del Fe un ruolo importante è svolto dalle perdite fisiologiche e dalle richieste dell’organismo in rapporto all’età e alla crescita. Nel maschio adulto la perdita giornaliera è di circa 1 mg, nella donna in età fertile la perdita è il doppio, poiché con l’emorragia mestruale sono perduti mediamente 30 mg di Fe. Dopo una gravidanza il bilancio netto del Fe comporta un deficit medio di 700 mg, quindi con una perdita giornaliera di poco meno di 3 mg. Dalla nascita fino ai 12 anni di età il fabbisogno giornaliero aumenta in ragione di 40 mg/Kg e dai 12 ai 18 anni nei maschi l’incremento della richiesta è di 0.4 mg/24 h, nelle femmine di 0.6 mg/24 h.
Gli altri due elementi indispensabili per una corretta maturazione degli eritroblasti sono la vit. B12 e l’acido folico o vit. B9. Ambedue queste vitamine agiscono sulla sintesi degli acidi nucleici. In particolare la vit. B9 subisce la trasformazione in tetraidrofolato (THF) tramite la di-idrofolico-ossidoreduttasi (DHFR). Il THF è la molecola accettrice del radicale idrossimetilico che per mezzo dell’omonima transferasi trasforma la serina in glicina; ma lo stesso gruppo può compiere il percorso inverso ad opera della serin-transidrossi-metilasi che produce serina dalla glicina, ovviamente estraendo il gruppo idrossimetilico dal metile-THF. L’ N5,N10-metilen-THF può subire la trasformazione in N5,N10-metenil-THF ad opera di un’ossidasi specifica. L’N5,N10-metenil-THF è il cofattore della transferasi specifica che sintetizza la 2’-deossitimidina-5’-fosfato dalla 2’-deossiuridina-5’-fosfato, precursore fondamentale della timina, base essenziale per la sintesi del DNA. L’N5,N10-metenil-THF può ciclizzare sulla posizione N5 o N10 e trasformarsi in N5- o N10-formil-THF per l’azione, rispettivamente, di una ciclo-idrolasi e di una sintetasi specifica. L’ N10-formil-THF svolge il ruolo fondamentale di coenzima in due tappe del ciclo di sintesi delle purine, basi essenziali sia per il DNA che per l’RNA. Quindi, è evidente l’assoluta necessità di THF metilato per sostenere un’attività metabolica di sintesi delle basi puriniche e pirimidiniche in quei tessuti, come il midollo osseo rosso, ad alto quoziente di cellule in fase proliferativa.
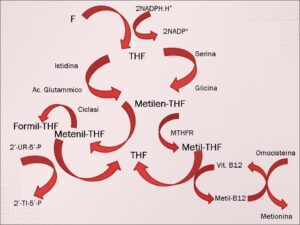
L’N5-metil-THF, che per la maggior parte è prodotto dalla conversione del gruppo metilenico tramite una reduttasi specifica FAD-dipendente, partecipa alla donazione del gruppo metilico all’omocisteina perché questa sia convertita in metionina ad opera della metionin-sintetasi. Il processo avviene in due tappe: il gruppo metilico è trasferito ad opera della metionin-sintetasi dalla vit.B12 all’omocisteina, ma la vit.B12 è metilata ad opera della metil-tetraidrofolato-ossidoreduttasi, la quale riconosce come coenzima l’N5-metil-THF. Questo crocevia vit. B12-THF all’altezza della sintesi della metionina comporta che la riduzione di concentrazione della vit.B12, finisce per accumulare metilen-THF e quindi finisce per diminuire la disponibilità di THF per ricevere i gruppi metilenici metenilici e metilici. Questo fenomeno metabolico sta alla base della così detta “trappola dei folati”, poiché livelli normali o in eccesso di vit.B9 rispetto alla vit.B12, come, appunto, nelle carenze di quest’ultima, comporta il mascheramento degli effetti deleteri che la carenza di vit.B12 ha sulla sintesi degli acidi nucleici [11][12].
La vit. B12 partecipa però ad un’ altra tappa metabolica: la conversione del meti-lmalonil-CoA in succinil-CoA ad opera della metil-malonil-CoA-mutasi. Il metil-malonil-CoA, a sua volta, deriva dal propionil-CoA carbossilato da un enzima specifico che ha come coenzima la vit.B7. Il propionil-CoA deriva dalla degradazione di alcuni aminoacidi (metionina, treonina, valina, isoleucina), del colesterolo e delle catene di acidi grassi ad atomi dispari. L’accumulo di metil-malonato all’interno dei mitocondri, dove avvengono tutte le reazioni di sintesi del succinato, provoca una forte riduzione dell’ α-chetoglutarato e dell’attività dell’ α-chetoglutarato-ossidoreduttasi, che riduce notevolmente l’attività del ciclo degli acidi tricarbossilici glutammato-dipendente, cosa che avviene soprattutto nelle cellule neuronali.
La vit. B9 è assunta generalmente complessata all’acido glutammico soprattutto come poliglutammato. La scissione avviene in forma di mono- o di-glutammato avviene a livello del digiuno, mentre l’assorbimento si attua prevalentemente nel tratto medio del grosso intestino. Tutta la vit. B9 coniugata al residuo mono- o di-glutammico è portata al fegato dove avvengono le reazioni metaboliche della DHFR, tappa necessaria affinchè la vitamina acquisisca la funzione di cofattore enzimatico. Il fabbisogno giornaliero in un adulto è di circa 400 μg, ma cresce in caso di gravidanza (600 μg) e di allattamento (500 μg), mentre nel neonato fino ad 1 anno di età è di circa 70 μg, in età scolare è di 200 μg ed è pari a quella dell’adulto a 20 anni [13].
La vit. B12, invece, è liberata dai complessi organici in cui è contenuta dall’acidità gastrica; in questa fase è protetta dalle Proteine R (PR), di origine salivare, dalla degradazione che potrebbe subire da parte della stessa acidità gastrica. Nel duodeno le proteasi pancreatiche rimuovono le PR mentre la vit. B12 di nuovo libera è complessata dal Fattore Intrinseco (FI), una proteina prodotta dalle cellule parietali dello stomaco. Giunta nel tratto terminale dell’Ileo il complesso FI-Vit.B12 è assorbito dalle cellule intestinali tramite un recettore specifico e la vit. B12 è trasferita al polo vascolare dell’enterocita da dove la transcobalamina II, una proteina carrier specifica, la trasporta tramite il circolo portale al fegato, poi da qui a tutti i tessuti. Questo meccanismo di assorbimento, piuttosto complesso, è molto inefficiente, perché della quantità ingerita è assorbito, al massimo, il 30%. La metà dell’intero quantitativo di vit. B12 stoccata nell’organismo, pari in media a 4 mg, è conservata nel fegato e da questo eliminata tramite la bile nell’intestino da dove è nuovamente riassorbita. Durante questo transito intestinale si perdono circa 3 μg al giorno di vit. B12, che è il reale fabbisogno giornaliero di un adulto. Il fabbisogno giornaliero in caso di gravidanza e allattamento aumenta di appena il 10%. Nel neonato il fabbisogno giornaliero è 1/6, mentre nell’età scolare sale alla metà per raggiungere la parità rispetto a quello dell’adulto a 20 anni [14].
Conclusioni: l’eritropoiesi consiste in un complesso processo maturativo a cui vanno incontro le cellule staminali midollari orientate in senso eritroide ad opera di fattori multipli: programma genetico differenziativo interno tramite attivazione sequenziale di fattori di trascrizione specifici per la sintesi dei tre principali elementi della differenziazione eritroide: la sintesi dell’emoglobina, la sintesi del recettore dell’eritropoietina e la sintesi del recettore della transferrina. Nel corso del processo differenziativo le cellule più immature perdono l’attività di autorinnovamento, presentano una progressiva degenerazione del nucleo, dei mitocondri, dei ribosomi, in maniera concomitante al progressivo accumulo di emoglobina. L’eritropoietina svolge in questo senso un ruolo fondamentale promuovendo la trascrizione dei geni differenziativi. La sintesi dell’emoglobina, che è il fenomeno metabolico preponderante durante le tappe maturative dell’eritroblasto, dipende esclusivamente dalla disponibilità di Fe, vit. B9, vit. B12. In particolare, la biodisponibilità del Fe dipende dal suo regolatore principale, l’epcidina,
Bibliografia
1 – Blank U, Karlsson G, Karlsson S. Signaling pathways governing stem-cell fate. Blood 2008;111:492-503 [full text]
2 – Tsiftsoglou AS, Vizirianakis IS, Strouboulis J. Erythropoiesis: Model systems, molecular regulators and developmental programs. IUBMB Life 2009;61:800-30 [full text]
3 – Migliaccio AR. Erythroblast enucleation. Haematologica 2010;95:1985-88 [full text]
4 – Chasis JA, Mohandas N. Erythroblastic islands: niches for erythropoiesis. Blood 2008;112:470-8 [full text]
5 – Fisher JW. Erytyhropoietin: Physiology and Pharmacology update. Exp Biol Med 2003;228:1-14 [full text]
6 – Forget BG, Hardison RC. The normal structure and regulation of human globin gene clusters. [full text]
7 – Peixeiro I, Silva AL, Romao L. Control of human β-globin mRNA stability and its impact on beta-thalassemia phenotype. Haematologica 2011;96:905-13 [full text]
8 – Layer G, Reichelt J, Jahn D, Heinz BW. Structure and function of enzymes in heme biosynthesis [full text]
9 – Ganz T, Nemeth E. Iron metabolism: interactions with normal and disordered erythropoiesis. Cold Spring Harb Perspect Med 2012;2(5):a011668 [full text]
10 – Finberg KE. Unraveling mechanisms regulating systemic iron homeostasis. Hematology 2011;n.1:532-7 [full text]
11 – Wagner C. Symposium on the subcellular compartmentation of folate metabolism. J Nutr 1996;126:12285-12345 [full text]
12 – Ragsdale SW. Catalysis of methyl group transfers involving tetrahydrofolate and B12. Vitam Horm 2008;79:293-324 [full text]
13 – Ohrvik VE, Witthof CM. Human folate bioavailability. Nutrients 2011;3(4):475-96 [full text]
14 – Quadros EV. Advances in the understanding of cobalamin assimilation and metabolism. Br J Haematol 2010;148:195-204 [full text]